
James GallagherCorresponsal de salud y ciencia
 BBC/Fergus Walsh
BBC/Fergus WalshUna de las enfermedades más cruel y más devastadoras, Huntington, ha sido tratada con éxito por primera vez, dicen los médicos.
La enfermedad atraviesa las familias, mata implacablemente las células cerebrales y se asemeja a una combinación de demencia, la enfermedad de Parkinson y las neuronas motoras.
Un equipo de investigación emocional se lloró cuando describieron cómo los datos muestran que la enfermedad se ralentizó en un 75% en los pacientes.
Significa que la disminución que normalmente esperaría en un año tomaría cuatro años después del tratamiento, dando a los pacientes décadas de “vida de buena calidad”, dijo la profesora Sarah Tabrizi a BBC News.
El nuevo tratamiento es un tipo de terapia génica administrada durante 12 a 18 horas de delicada cirugía cerebral.
Los primeros síntomas de la enfermedad de Huntington tienden a aparecer en sus 30 o 40 años y normalmente son fatales en dos décadas, abriendo la posibilidad de que el tratamiento más temprano pueda evitar que surjan síntomas.
El profesor Tabrizi, director del Centro de Enfermedades del University College London Huntington, describió los resultados como “espectaculares”.
“Nunca en nuestros sueños más salvajes hubiéramos esperado una desaceleración del 75% de la progresión clínica”, dijo.
Ninguno de los pacientes que han sido tratados están siendo identificados, pero uno fue retirado médicamente y ha regresado al trabajo. Otros en el juicio todavía están caminando a pesar de que se espera que necesiten una silla de ruedas.
Es probable que el tratamiento sea muy costoso. Sin embargo, este es un momento de verdadera esperanza en una enfermedad que golpea a las personas en su mejor momento y devasta a las familias.

 BBC/Fergus Walsh
BBC/Fergus WalshHuntington corre por la familia de Jack May-Davis. Tiene el gen defectuoso que causa la enfermedad, al igual que su padre, Fred y su abuela, Joyce.
Jack dijo que fue “realmente horrible y horrible” ver el inexorable declive de su padre.
Los primeros síntomas aparecieron a finales de los años 30 de Fred, incluidos los cambios en el comportamiento y la forma en que se movió. Finalmente necesitaba cuidados paliativos las 24 horas, los 7 días de la semana, antes de morir a la edad de 54 años, en 2016.
Jack tiene 30 años, el empleado de un abogado, recién comprometido con Chloe y ha participado en la investigación en UCL para convertir su diagnóstico en positivo.
Pero siempre había sabido que estaba destinado a compartir el destino de su padre, hasta hoy.
Ahora dice que el “avance” absolutamente increíble “lo ha dejado” abrumado “y capaz de mirar hacia un futuro que” parece un poco más brillante, me permite pensar que mi vida podría ser mucho más larga “.
 May-Davis Family
May-Davis FamilyLa enfermedad de Huntington es causada por un error en parte de nuestro ADN llamado gen Huntingtin.
Si uno de sus padres tiene la enfermedad de Huntington, existe un 50% de posibilidades de que heredes el gen alterado y eventualmente desarrollarás Huntington.
Esta mutación convierte una proteína normal necesaria en el cerebro, llamada proteína de huntingtina, en un asesino de neuronas.
El objetivo del tratamiento es reducir los niveles de esta proteína tóxica permanentemente, en una sola dosis.
La terapia utiliza medicina genética de vanguardia que combina terapia génica y tecnologías de silenciamiento génico.
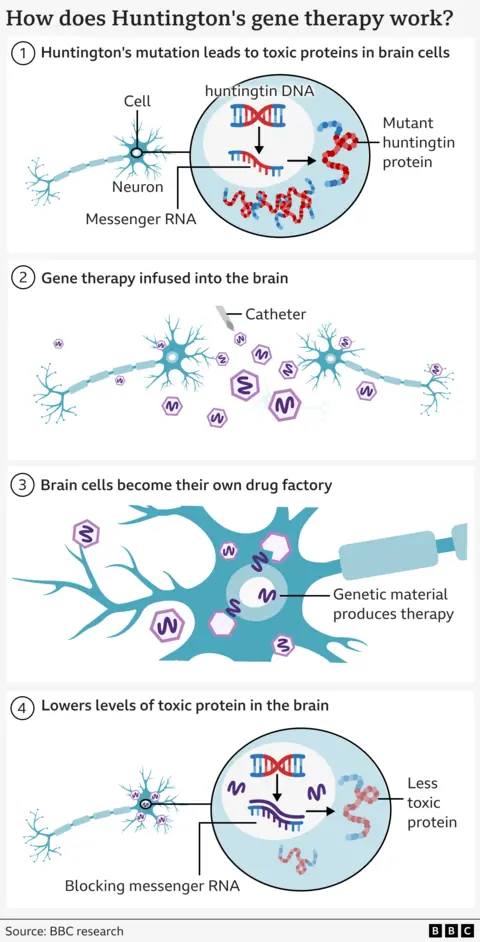
Comienza con un virus seguro que se ha alterado para contener una secuencia de ADN especialmente diseñada.
Esto se infunde profundamente en el cerebro utilizando una exploración por resonancia magnética en tiempo real para guiar un microcatéter a dos regiones cerebrales: el núcleo caudado y el putamen. Esto lleva de 12 a 18 horas de neurocirugía.
El virus luego actúa como un cartero microscópico, que entrega la nueva pieza de ADN dentro de las células cerebrales, donde se activa.
Esto convierte las neuronas en una fábrica para hacer la terapia para evitar su propia muerte.
Las células producen un pequeño fragmento de material genético (llamado microARN) que está diseñado para interceptar y deshabilitar las instrucciones (llamadas ARN mensajero) que se envían desde el ADN de las células para construir huntingtina mutante.
Esto da como resultado niveles más bajos de caza mutante en el cerebro.
 UCLH
UCLHLos resultados del ensayo, que involucró a 29 pacientes, han sido emitidos en un comunicado por la compañía Uniqure, pero aún no han sido publicados en su totalidad para su revisión por otros especialistas.
Los datos mostraron que tres años después de la cirugía hubo una desaceleración promedio del 75% de la enfermedad en función de una medida que combina la cognición, la función motora y la capacidad de manejar en la vida diaria.
Los datos también muestran que el tratamiento está guardando células cerebrales. Los niveles de neurofilamentos en el líquido espinal, un signo claro de las células cerebrales, deberían haber aumentado en un tercio si la enfermedad continuó progresando, pero en realidad era más bajo que al comienzo del ensayo.
“Este es el resultado que hemos estado esperando”, dijo el profesor Wild, neurólogo consultor del Hospital Nacional de Neurología y Neurocirugía de UCLH.
“Existían la posibilidad de que nunca veríamos un resultado como este, por lo que vivir en un mundo donde sabemos que esto no solo es posible, sino que la magnitud real del efecto es impresionante, es muy difícil encapsular completamente la emoción”.
Dijo que estaba “un poco lloroso” pensando en el impacto que podría tener en las familias.
El tratamiento se consideró seguro, aunque algunos pacientes desarrollaron inflamación del virus que causó dolores de cabeza y confusión que resolvieron o necesitaban tratamiento con esteroides.
El profesor Wild anticipa que la terapia “debería durar vida” porque las células cerebrales no son reemplazadas por el cuerpo de la misma manera que la sangre, el hueso y la piel se renovan constantemente.
Aproximadamente 75,000 personas tienen la enfermedad de Huntington en el Reino Unido, EE. UU. Y Europa, con cientos de miles que llevan la mutación, lo que significa que desarrollarán la enfermedad.
Uniqure dice que solicitará una licencia en los EE. UU. En el primer trimestre de 2026 con el objetivo de lanzar el medicamento más tarde ese año. Las conversaciones con las autoridades en el Reino Unido y Europa comenzarán el próximo año, pero el enfoque inicial está en los Estados Unidos.
El Dr. Walid Abi-Saab, director médico de Uniqure, dijo que estaba “increíblemente emocionado” sobre lo que significan los resultados para las familias, y agregó que el tratamiento tenía “el potencial de transformar fundamentalmente” la enfermedad de Huntington.
Sin embargo, el medicamento no estará disponible para todos debido a la cirugía altamente compleja y al costo anticipado.
“Será costoso seguro”, dice el profesor Wild.
No hay un precio oficial para el medicamento. Las terapias genéticas a menudo son caras, pero su impacto a largo plazo significa que aún puede ser asequible. En el Reino Unido, el NHS paga una terapia génica de £ 2.6 millones por paciente para la hemofilia B.
El profesor Tabrizi dice que esta terapia génica “es el comienzo” y abrirá las puertas de terapias que pueden llegar a más personas.
Ella rindió homenaje a los voluntarios “verdaderamente valientes” que participaron en el juicio, diciendo que estaba “encantada con los pacientes y las familias”.
Ella ya está trabajando con un grupo de jóvenes que saben que tienen el gen, pero aún no tiene síntomas, conocidos como Stage Zero Huntington’s, y tiene como objetivo hacer el primer ensayo de prevención para ver si la enfermedad puede retrasarse significativamente o incluso detenerse por completo.








{kind=link}